What is LQTS?
- Long Q-T syndrome is a disorder of the heart’s electrical system.
- The electrical activity of the heart is produced by the flow of ions (electrically charged particles of sodium, calcium, potassium, and chloride) in and out of the cells of the heart. Tiny ion channels control this flow.
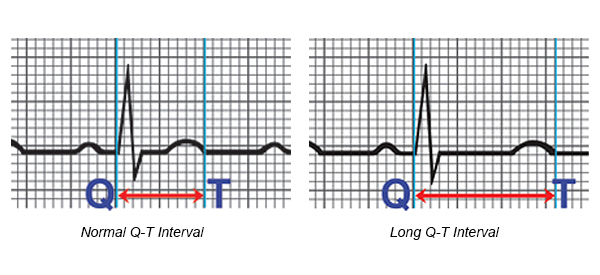
- The Q-T interval is the section on the electrocardiogram (ECG) - that represents the time it takes for the electrical system to fire an impulse through the ventricles and then recharge. It is translated to the time it takes for the heart muscle to contract and then recover.
- LQTS occurs as the result of a defect in the ion channels, causing a delay in the time it takes for the electrical system to recharge after each heartbeat. When the Q-T interval is longer than normal, it increases the risk for torsade de pointes, a life-threatening form of ventricular tachycardia.
- LQTS is rare. The prevalence is about 1 in 5,000 persons in the United States.
What causes LQTS?
Long Q-T syndrome can be acquired or congenital:Acquired LQTS is caused by many medications. Sensitivity to these medications may be related to genetic causes.
Congenital LQTS is usually inherited. It is caused by an abnormality in the gene code for the ion channels. The abnormality of the ion channels slows the recovery phase of the heartbeat. Forms of inherited LQTS include:
- Recent Classifications – Multiple ion channel abnormalities have been discovered. The most common ones include LQT1, LQT2, LQT3, LQT4, LQT5; these are classified by the type of channel which causes the LQTS. The type of LQTS classification is related to the risk of future cardiac events, those with LQT3 having the highest risk of life-threatening arrhythmias.
- Jervell, Lange-Nielsen Syndrome (autosomal recessive inheritance pattern) – Both parents are carriers of the abnormal gene, but they may not manifest LQTS. Each child has a 25-percent chance of inheriting LQTS. This syndrome is associated with deafness at birth and is extremely rare, as there is a small chance that both parents would carry the LQTS gene.
- Romano-Ward Syndrome (autosomal dominant inheritance pattern) – One parent has LQTS and the other parent usually does not. Each child has a 50-percent chance of inheriting the abnormal gene. In this syndrome, hearing is normal; however the likelihood that children in this family would have LQTS is greater. The gene may be present in all the couple’s children, some of them or none at all.
- Children who are deaf at birth
- Children and young adults who have unexplained sudden death or syncope in family members
- Blood relatives of family members with LQTS
- Those with LQTS taking medications that can further prolong the QT intervals. See medications to avoid under Treatment Options.
Do You Need to be Screened for Long QT Syndrome?
Long QT Syndrome is a medical condition that can be passed on from generation to generation. It is important for you to be screened for this condition if you have a first-degree relative with Long QT Syndrome. First-degree relatives are your parents, siblings and children.The first step is to tell your doctor that you have a family history of this condition. He or she may want to do diagnostic tests to check your heart. If these tests are positive, you should be seen by a cardiologist who is familiar with this condition.
What are the symptoms?
The most common symptoms include:- Syncope (fainting)
- Seizures
- Sudden death
Symptoms are most common during:
- Exercise (or within a few minutes after)
- Emotional excitement, especially being startled
- During sleep or upon waking suddenly
How is LQTS diagnosed?
LQTS is usually diagnosed by measuring the Q-T interval on the ECG. Other testing may include:- Exercise stress test
- Ambulatory monitor
- Family history of LQTS
- Family history of unexplained fainting, seizures, or cardiac arrest
- History of fainting, seizures or cardiac arrest, especially with exercise
How is it treated?
Treatment is aimed at preventing sudden death and controlling symptoms. Treatment includes:Medications
Most patients (even those without symptoms) are treated with a beta-blocker. Other medications may be used to shorten the Q-T interval. Your doctor will discuss what medications are best for you. It is important to know:- The names of your medications
- What they are for
- How often and at what times to take them
There are many medications that can prolong the QT interval. Those with LQTS may be more prone to the effects of these medications. If you have LQTS, you should:
- Do not take over-the-counter medications (except for plain aspirin or acetaminophen) without first talking to your health care provider.
- Tell all your health care providers you have LQTS, as there are many drugs you cannot take.
- Talk to your doctor before taking any medications prescribed for other medical conditions. The following types of medications may affect you if you have LQTS:
- Antihistamines
- Antidepressants, mental illness medications
- Heart medications
- Antibiotics, antifungals, antivirals
- Intestinal medications
- Anticonvulsants
- Diuretics
- Antihypertensives
- Migraine medications
- Cholesterol lowering medications
Devices
- Patients who have a history of cardiac arrest or symptoms, in spite of beta-blocker therapy, may receive an implantable cardioverter defibrillator (ICD). This device detects life-threatening arrhythmias and automatically shocks the heart to prevent sudden death.
- Patients who have an abnormally slow heart rate may receive a pacemaker.
Lifestyle changes
- Family testing - All first-line relatives (brothers, sisters, parents and children) should have EKG testing. Any other family members who have a history of seizures or fainting should also undergo testing.
- Exercise - If you have LQTS, sometimes, fatal arrhythmias occur with exercise. The decision to participate in competitive sports should be managed by a heart rhythm expert and certain precautions may be suggested.
- Buddy system - Your family and friends should be told you have LQTS. They should be told to call for emergency help (911) if you begin to have symptoms or faint.
This information is provided by the Cleveland Clinic and is not intended to replace the medical advice of your doctor or healthcare provider. Please consult your healthcare provider for advice about a specific medical condition.


No comments:
Post a Comment